




ProteinDFシステム
ProteinDF[1- 3]はタンパク質の精密なカノニカル全電子波動関数を計算するための密度汎関数法[4]に基づく分子軌道法プログラムです。オブジェクト指向言語C++ [5]によるプログラム構築技術を取り入れるとともに[1]、プログラムを並列化し[2]、2001年に世界最大の金属タンパク質シトクロム c の全電子計算に成功[3]、2004年には306残基のインスリン6量体の全電子計算に成功した実績があります[6]。
タンパク質のさまざまな理論計算が進展し、ポストゲノム時代が花開こうとしています。この時代にProteinDFは科学技術、産業技術の両面で貢献できるプログラムの1つであると考えております。そのような観点から、ProteinDFを広く研究者、技術者に利用していただこうと整備を行っており、基本的な機能が使用できる バージョンが公開されております。
このProteinDFシステムでは、密度汎関数法による超大規模タンパク質全電子計算エンジンProteinDFに加え、ペプチド鎖からなるタンパク質の計算に特化したタンパク質全電子半自動計算法[7]と、タンパク質の構造最適化、分子動力学、自由エネルギー計算を行うProteinMD、そして、それらシミュレーションを支援し、物性を評価・解析できる高品位GUI(グラフィカルユーザインターフェース)プログラム(ProteinEditor) を搭載いたしました。このような系であれば、すぐにでも計算を実行に移すことができます。
我々はこのProteinDFをさらに進化させて参ります。ユーザの皆さまの声を反映し、より使いやすく、よりさまざまなマシンに対応し、より機能の豊富なプログラムへと発展させていく所存ですので、今後とも忌憚の無いご意見、ならびにご協力をお願いいたします。
タンパク質全電子半自動計算法(QCLO法)
QCLO法[7]はタンパク質の全電子計算を簡単に、安全に且つほぼ自動的に達成するために当グループで開発された手法です。タンパク質は、量子化学計算を行う分子としては、非常に巨大で複雑な分子です。そのため、タンパク質の全電子計算は、通常分子の計算とは異なり、専門家をもってしても数々の試行錯誤を必要とし、一般ユーザが容易に達成できる計算ではありませんでした。そこで、このような難しいタンパク質の全電子計算をほとんど試行錯誤することなく達成する方法として、QCLO法を開発しました。
具体的には、分子軌道法の支配方程式は自己無撞着(SCF)法で解かれますが、タンパク質の電子状態は複雑で、大変よい初期値を用いないと全電子計算が達成できません。本グループは一種の局在化軌道(擬カノニカル局在化軌道:QCLO)を用いて初期値を作成する方法を開発しました。QCLOはアミノ酸残基単位で局在化しているが、その中ではアミノ酸残基単体の軌道であるカノニカル軌道に近い分子軌道のことです。これらを切り貼りすると、ペプチド鎖の分子軌道計算の大変よい初期値になることを発見しました(図1)。そこで、ペプチド鎖の分子軌道計算結果から、再度QCLOを求め、さらに大きなペプチド鎖の計算の初期値とし、これらのペプチド延長過程を繰り返せば、安全かつほぼ自動的にタンパク質の全電子計算が達成できるわけです。これを、QCLO法を用いたタンパク質全電子計算収束過程法と呼びます(図2)。これにより、初期値の誤差を従来の数十分の一にすることに成功しました。ProteinDFシステムには、これを自動的に実行する機能が組み込まれており、ほぼ全ての全電子計算を半自動計算する仕組みを提供しております。また、その高速計算システムを開発し、一般ユーザが容易に且つ高速にタンパク質の精密な量子化学計算を実行できるようになっています。
真の自動化への鍵は、ペプチド延長過程のシナリオが握っています。現在のペプチド延長過程は、どのようなタンパク質においても1残基から3残基、7残基・・・と機械的に延長していきます。しかし、タンパク質は複雑な立体構造を持っています。たとえアミノ酸残基の通し番号が離れていても、塩橋や水素結合を形成するなど強い相互作用を示す場合が多々あります。真の自動化を達成するためには、このような相互作用を考慮に入れた計算を行う必要があります。そのため、立体構造情報を元にタンパク質の全電子計算を達成させるQCLO法の開発も進めております[8]。これにより、より安全かつ自動的なタンパク質の全電子計算の達成を予定しております。
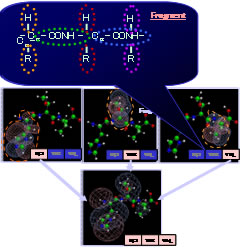
図 1 QCLOによる初期値作成の概念図
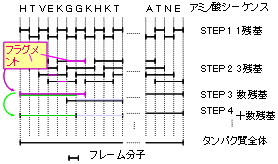
図 2 QCLO法を用いたタンパク質全電子計算収束過程法
ProteinMD
ProteinMDは超大規模タンパク質全電子計算エンジンProteinDF[1-3]を中心としたProteinDFシステムの構成エンジンの1つです。このシステム内でタンパク質の構造最適化、分子動力学法シミュレーション、自由エネルギー計算等を実行することができます。タンパク質の全電子波動関数計算を実行するには、そもそも計算対象であるタンパク質の立体構造が必要です。タンパク質の立体構造はProtein Data Bank(PDB)[9]から入手が可能ですが、必ずしもその構造が適切なものである保証はありません。むしろ、異常な歪みを持つことが多く、それらを修正する必要があります。また、PDBなどに登録されていないようなタンパク質の場合、何らかの方法でその適切な立体構造を得る必要があります。これらの問題の解決策の第1歩として経験的な分子力場を用いた構造最適化が有効です。このような全電子波動関数計算のプレ処理機能を提供します。
生体内のタンパク質は300Kほどの温度の水溶液中に存在し、絶えずその構造は揺らいでいます(図3)。また、僅かな溶媒の変化に対してその構造は適宜に変化し、それぞれのタンパク質は機能を活性化させています。実際のタンパク質は溶媒中での反応の連続といえます。そのようなタンパク質の特性を知る上で、それらのダイナミクスの解析は必須です。タンパク質のダイナミクスの解析において、分子動力学法シミュレーションは最も有効であり、その機能を提供します。
実験ではタンパク質の一瞬の構造(状態)のみを観測しているわけではありません。様々な状態の熱平均を観測しています。そのため、実験との比較には統計力学に則った理論計算が必要です。タンパク質の溶媒和エネルギーや、構造の変化によるエネルギー変化、さらに、分子間距離などに対する平均力ポテンシャルなどの自由エネルギーを正確に見積もるには様々な状態の十分なサンプリングが必須となります。この十分なサンプリング計算を達成するにはシミュレーション計算の高速化や先駆的なサンプリング・アルゴリズムが不可欠です。ProteinMDでは、実用的なタンパク質の解析機能を提供すべく、プログラムの改善・開発が為されています。
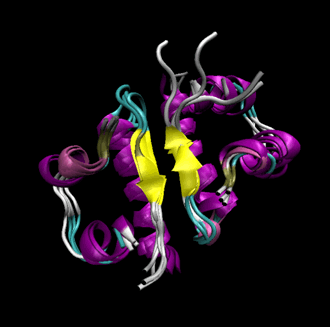
図 3 インスリン・アナログ、アスパルト。生体内ではタンパク質の構造は揺らいでおり、分子動力学法はそのような系の研究に有効である。
タンパク質波動関数データベース
ProteinDFシステムによる全電子計算の結果から、タンパク質の様々な物理量や知見を得ることができます。こうしたタンパク質の情報は、医薬、触媒、食料、環境・エネルギー分野に応用することができます。有益な情報が得られるタンパク質の全電子計算ですが、一方で大規模な計算資源と長時間の計算時間も必要です。そこでタンパク質全電子計算の結果を蓄積し、効率良い計算と結果の再利用する仕組みが期待されます。当研究室では、系統的にタンパク質の全電子計算を行い、タンパク質波動関数データベースを作成しました。このデータベースはインターネットなどを通じて公開することにより、人類共通の貴重な財産となるでしょう。タンパク質全電子計算の結果を随時追加し、タンパク質波動関数データベースを更新しています。
タンパク質の全電子計算によって得られた結果から、様々な物理量を計算することができます。図4に示した、分子軌道図や静電ポテンシャル分布図などは、ほんの一例です。タンパク質全電子計算の結果から求められるデータは、医薬、触媒、食料、環境・エネルギー分野に応用できる、非常に貴重なデータです。しかし、タンパク質の全電子計算には、CPUやメモリなど膨大な計算機資源と時間が必要であり、現在誰もが気軽に計算することは困難な状況にあります。
そこで、タンパク質全電子計算によって得られた結果やデータを蓄積・管理する仕組みが求められます。このデータベースをCD/DVDやインターネットなどで公開することにより、誰もが自由にタンパク質全電子計算の結果にアクセスすることができ、また計算した結果をデータベースに登録して公開することもできます。様々な種類のタンパク質のデータを数多く登録して、縦横に検索・解析できれば、そこから新しい発見が見出される可能性もあります。
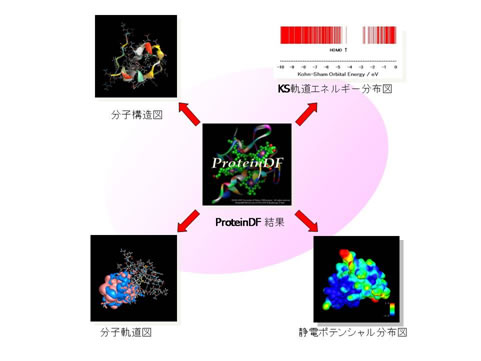
図 4 タンパク質全電子計算から得られる様々な物理量
ProteinEditor
統合環境として開発されたProteinEditorは、量子化学計算に関わる研究者のみならず、生物物理学や生化学の研究者を想定して開発され、現在ではProteinDFによる複雑なタンパク質の全電子計算達成をサポートする統合環境としての機能をほぼ全て装備しています。システムを統括する機能に加えてグラフィカルな編集機能、各種の機能表現に必要なタンパク質のための大規模分子グラフィックス、およびそのGUIで構成されています。
ProteinEditorはWindows上で動作するアプリケーションで、分子グラフィックスにはOpenGLを用い、高速な分子モデルの描画を実現しています。Protein Data Bank (PDB)が配信する標準タンパク質立体構造データを表示し、解析する機能が充実しており、Ramachandranプロット機能、結合長・結合角・二面角の分布表示機能、異常原子間距離表示機能によってタンパク質の構造妥当性の評価が行いやすくなっています。また、アミノ酸置換(ミューテーション)機能、簡易MD組み込みによる構造緩和機能、MDアニメーション表示機能なども組み込まれていて簡単なモデリングも行えるようになっています。処々の機能は様々な3次元グラフィックスと連動しているので、非常に扱いやすいGUIになっています。
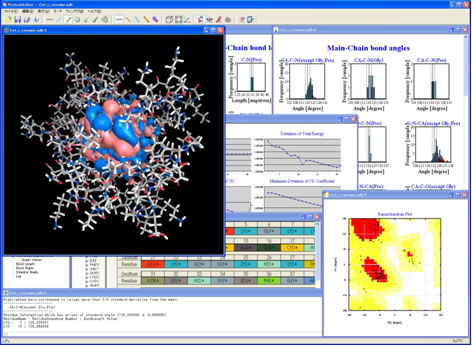
図 5 ProteinEditorのスナップショット
参考文献
- 1.
- F. Sato, Y. Shigemitsu, I. Okazaki, S. Yahiro, M. Fukue, S. Kozuru, H. Kashiwagi, Int. J. Quant. Chem., 63 (1997) 245.
- 2.
- T. Yoshihiro, F. Sato, H. Kashiwagi, Chem. Phys. Lett., 346 (2001) 313.
- 3.
- F. Sato, T. Yoshihiro, M. Era, H. Kashiwagi, Chem. Phys. Lett., 341 (2001) 645.
- 4.
- W. Kohn, L. J. Sham, Phys. Rev., 140 (1965) A1133.
- 5.
- B. Stroustrup: “The C++ Programing Language(Special Edition)”, Addison Wesley. Reading Mass. USA. (2000).
- 6.
- T. Inaba, H. Tsunekawa, T. Hirano, T. Yoshihiro, H. Kashiwagi, F. Sato, Chem. Phys. Lett., 434 (2007) 331.
- 7.
- H. Kashiwagi, H. Iwai, K. Tokieda, M. Era, T. Sumita, T. Yoshihiro, F. Sato, J. Mol. Phys. 101 (2003) 81.
- 8.
- N. Nishino, T. Hirano and F. Sato, 投稿準備中
- 9.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, P. E. Bourne, The Protein Data Bank, Nucleic Acids Research, 28 (2000) 235.