|
現在、タンパク質やDNAなどの生体高分子を原子レベルで取り扱うシミュレーションはAMBERやCHARMMのようなデータベース型の力場を利用したものが主流です。シミュレーション結果の精度に最も大きく影響するのは力場(モデル)であり、シミュレーションの正確さは、採用している力場がどれほど正しいものであるかによります。確かに、これらの力場はアミノ酸残基の種類や原子の種類によって値が異なるよう注意深く作成されているようです。しかし、問題は、これらのモデルではタンパク質内の位置に依らず、同じアミノ酸残基であればまったく同じパラメータ値を利用することです。
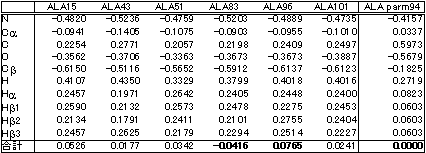
表にシトクロムcの全電子計算によるアラニンのMulliken電荷と参考のためにAMBER94のparmのアラニンの点電荷を示してあります。もちろん、両者の定義は異なりますので、これらの間で直接値を比較することはできません。しかし、全電子計算による結果を見てみますと、同じアラニンでもタンパク質内の位置によって電荷が異なることが分かります。しかも、その違いは83番目と96番目のアラニンの差で0.1ほどもあります。電荷の0.1の違いが10Å先に与える影響はほぼ0.1eV(≒3kcal/mol)にもなります。生体内で働くタンパク質は、自由エネルギー変化が0.1eV程のわずかな変化で構造や機能を変化させているわけですから、この違いは無視できません。アミノ酸単位でパラメータを構築するモデルでは、アミノ酸全体の電荷の合計が強制的に整数値(この場合中性なので0)を持たざるを得ないので、この違いを考慮に入れることは原理的にできません。結局、より正確な力場を用いる必要があり、タンパク質の全電子計算は明瞭かつ唯一の解決策ではないでしょうか。
|